




合作客戶/
拜耳公司 |
同濟大學 |
聯合大學 |
美國保潔 |
美國強生 |
瑞士羅氏 |






相關新聞Info
水和乙二醇-水混合體系中的離子液體-陽離子表面活性劑混合膠束自聚焦-電導法 表面張立法和光譜研究法—
來源:上海謂載 瀏覽 1746 次 發布時間:2021-12-16
3.結果和討論
3.1.臨界膠束濃度與膠束性質
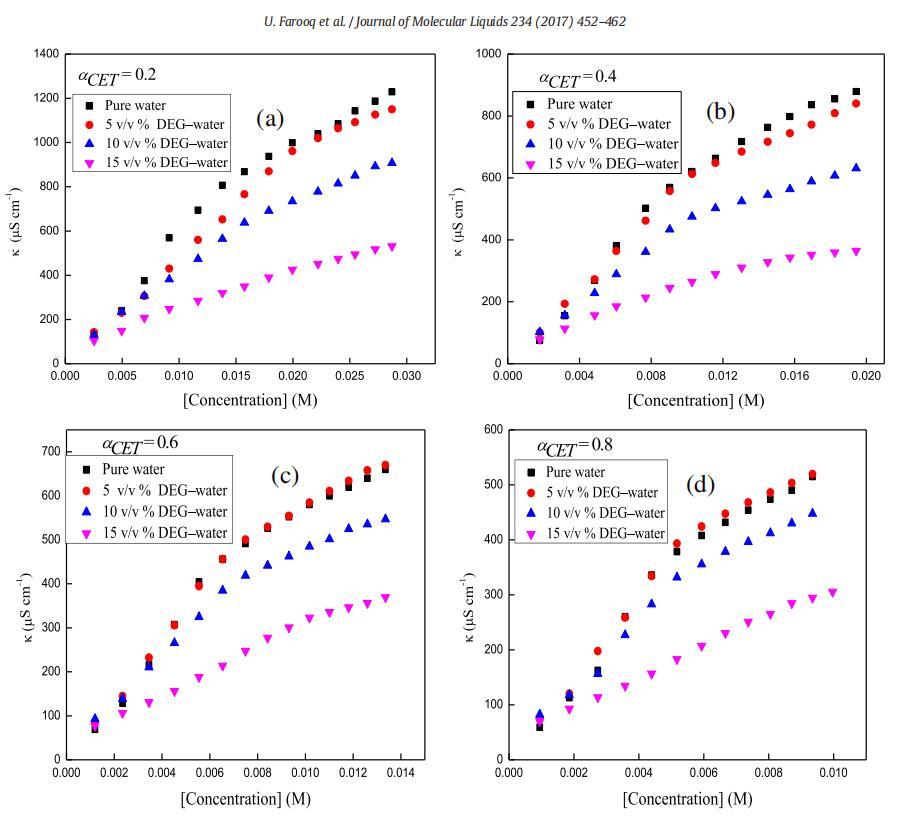
圖1。在T=298.15 K時,二甘醇-水二元混合物的不同體積百分比下,混合體系的比電導率,κ與[濃度]的曲線圖,CET+[C10min][Cl](a)αCET=0.2(b)αCET=0.4(c)αCET=0.6和(d)αCET=0.8。
在低濃度下,兩親分子表現為簡單電解質,并遵循Onsager方程[58,59],然而,電導率在一定濃度以上顯著降低,且不遵守Onsager方程。通過電導和表面張力技術測定了純表面活性劑(CET)、純離子液體[C10min][Cl]和混合物CET+[C10min][Cl]在水中以及不同體積百分比的二甘醇-水二元溶劑中的cmc值,如圖所示。1和2。表1總結了純水中CET、[C10min][Cl]和CET+[C10min][Cl]混合系統以及存在不同體積百分比的二甘醇-水二元混合物時的cmc值。通過張量法進一步確認電導率計算的cmc值,以進一步檢查值的準確性。純CET[37]和純[C10min][Cl][38]的文獻值與實驗值(表1)吻合良好。通過表面張力法和電導率法獲得的cmc值彼此不同,表明cmc是一個依賴于方法的參數。由于從兩種技術獲得的cmc值相差很小,因此可以很容易地求出平均值[39]。在水介質中,膠束化過程主要取決于表面活性劑頭基團之間的靜電相互作用和表面活性劑分子烴鏈之間的疏水相互作用[39]。然而,在非水介質中,烴鏈的疏溶劑性是表面活性劑分子膠束化的主要驅動力[40]。本文研究了陽離子表面活性劑和陽離子離子液體,它們的頭基都帶正電荷,因此靜電相互作用不利于膠束化,從表1可以看出,混合系統CET+[C10min][Cl]在不同CET摩爾分數下的cmc值介于各個值之間。這可能是由于疏水相互作用的增加,從而克服了表面活性劑分子之間的靜電相互作用。隨著有機共溶劑用量的增加,膠束化過程延遲[41–43]。DEG的介電常數為29.11[44],低于水的介電常數78.21[45],溶劑介電常數的降低可能導致表面活性劑分子帶電頭基團之間的排斥作用增加,并可能導致cmc值增加。隨著CET(αCET)摩爾分數的增加,混合系統的cmc降低,并且這種降低在水介質中比在二甘醇-水二元混合物中更大(圖3)。這種減少可能是由于疏水性增加,或者換句話說,表面活性劑單體疏水基團之間的疏水-疏水相互作用增加,導致早期膠束形成,因此cmc值降低。此外,隨著混合系統中DEG體積百分比的增加,介質的介電常數降低,這導致正電荷頭基團之間的排斥作用增強,因此觀察到cmc值增加。不同的工作人員使用自然共振理論(NRT)[46]對咪唑環進行了自然鍵軌道(NBO)分析,從他們的研究中發現,C2\H鍵的NBO值降低清楚地表明[C10min]+的氫質子與十六烷基三胺的氮基團之間存在氫鍵,在C2氫和Br之間?離子,以及溶劑分子和[C10min]+[47]的C2氫之間。
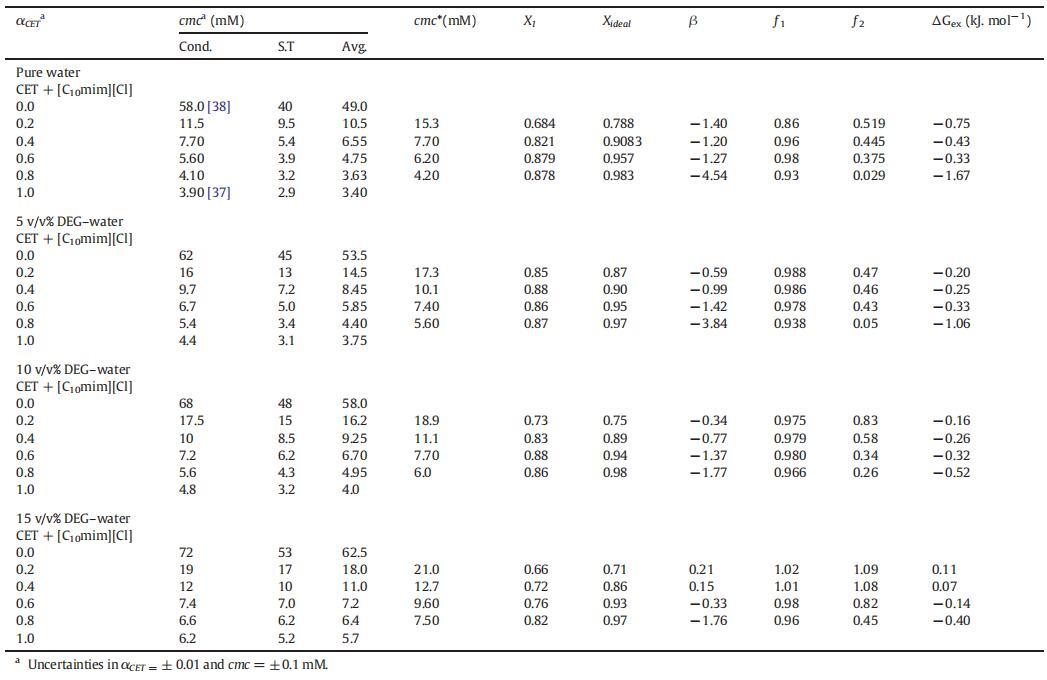
表1(cmc)、平均值(cmc)、理想值(cmc*)、膠束組成(X1,Xideal)、相互作用參數(β)的值,混合物CET+[C10min][Cl]的活度系數(?1,?2)和混合過量自由能(ΔGex),作為純水中CET摩爾分數(α)的函數,以及在T=298.15 K時DEG-水二元混合物的不同體積百分比。
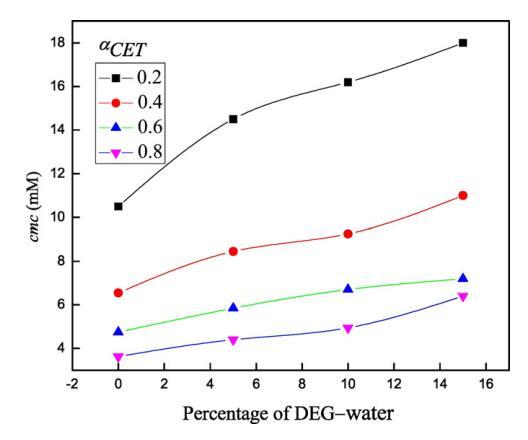
圖3。不同DEG-水混合物體積百分比下cmc的變化(a)αCET=0.2(■),(b)在298.15 K時,αCET=0.4(),(c)αCET=0.6()和(d)αCET=0.8()。
應用正規溶液理論,可以計算混合膠束中表面活性劑分子之間的相互作用參數(β)值,從而提供表面活性劑分子之間相互作用強度的信息。借助正規溶液理論(RST),膠束摩爾分數也可以使用魯賓方程計算:
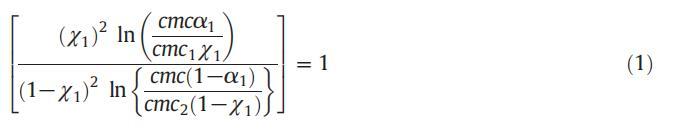
式中,cmc1、cmc2和cmc分別表示CET、[C10min][Cl]及其混合體系的實驗cmc值,χ1表示表面活性劑的膠束摩爾分數(CET),α1表示組分1的體積摩爾分數。
在理想狀態下,膠束摩爾分數(χ理想)已通過公式(2)[50]計算得出:
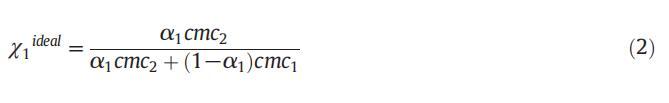
由β表示的相互作用參數給出了混合膠束中兩個單體之間的吸引相互作用的信息,可使用式(3)計算:

表1列出了根據式(3)計算的純水中的β值。在存在不同體積百分比的二甘醇-水二元溶液的情況下,也測定了類似值,如表1所示。β的值可以是正(拮抗作用)、負(協同作用)或零(組分之間無相互作用)[51]。此外,β的負值意味著IL和表面活性劑之間的吸引力大于單個組分之間的吸引力。從表1可以看出,對于所有二元混合物,β值均為負值,然而,隨著DEG的量從5增加到15 v/v%,負值減小,令人驚訝的是,對于αCET=0.2和0.4,β值均為正值。β的負值通常是混合膠束中有利相互作用的原因,然而正值意味著不利相互作用[53–54]。因此,從表1中可以看出,作為膠束形成主要力量的協同作用隨著DEG量的增加而減少。這種下降的原因可能是DEG的結構破壞能力導致單體之間的疏水相互作用減少,從而降低了它們的協同作用。
應用偽相模型,我們可以計算二元混合物的理想cmc,該模型基于這樣的假設,即膠束被視為宏觀相,與溶液中相應的單體處于平衡狀態。理想cmc值與單個cmc相關,可使用Clint公式(4)計算:

其中,cmc1和cmc2是組分1(CET)和組分2[C10min][Cl]的cmc,cmc*是混合物的理想cmc,α1是組分1在本體中的摩爾分數。如表1所示,實驗cmc值偏離理想cmc值,這表明混合二元系統中的非理想性。發現不同CET摩爾分數的CET+[C10min][Cl]混合系統的實驗cmc值較低,與基于理想混合的Clint模型預測的理想cmc*值呈負偏差,從而也證實了混合系統的非理想行為(見圖4)。
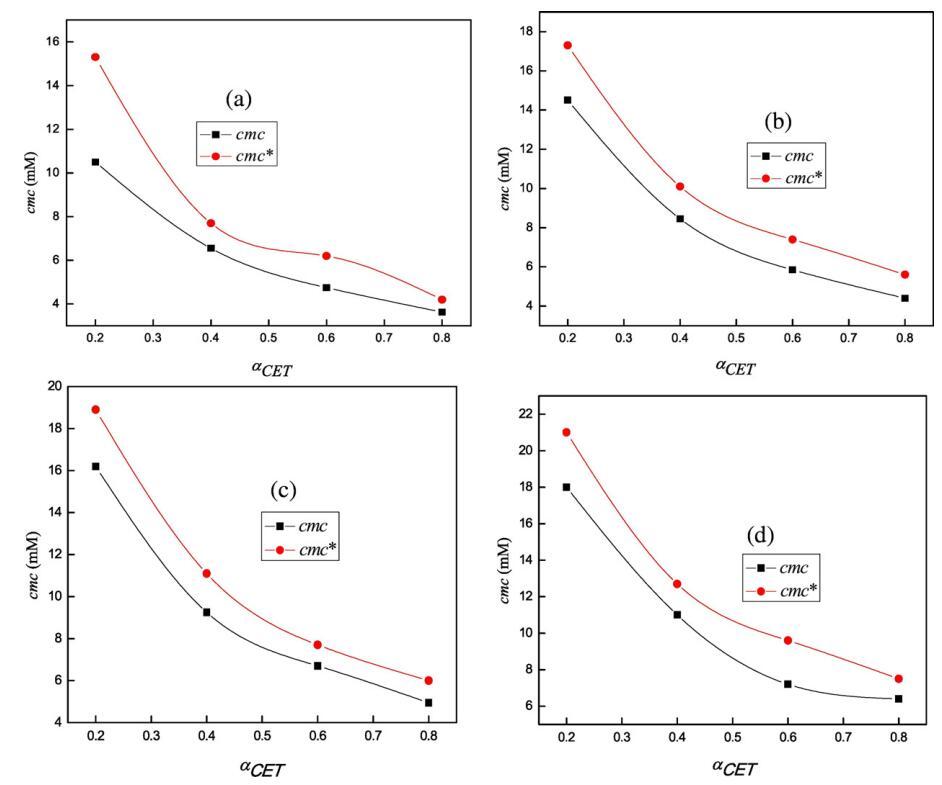
圖4。cmc的變化(■)在298.15 K條件下,在不同體積百分比的二甘醇-水混合物(a)存在于純水中(b)5 v/v%二甘醇-水(c)10 v/v%二甘醇-水和(d)15 v/v%二甘醇-水中時,使用αCET的cmc*()。
使用魯賓模型獲得的膠束摩爾分數(X1)用于計算混合膠束中[C10min][Cl]的CET活度系數?1和?2。(5)及(六):

如表1所列,大多數摩爾分數的活度系數?1和?2的值小于1,這表明混合系統具有吸引力的相互作用和非理想行為,但在αCET=0.2和0.4的CET+[C10min][Cl]混合物的情況下,出現了一些例外,其中,?1和?2的值大于1,表明混合膠束的形成不利,膠束化過程存在延遲。
活度系數以及膠束摩爾分數也可用于使用式(7)[56]計算混合的過量自由能(ΔGex):

表1所示的不同CET摩爾分數下混合系統的負ΔGex值為負值;這證實了混合膠束在熱力學上比單個組分的膠束更穩定。然而,αCET=0.2和0.4時的ΔGex值為正值(表1),這進一步支持了在這兩個摩爾分數下膠束化過程延遲的事實。
膠束化的標準吉布斯自由能可使用式(8)計算:

式中,χcmc是表面活性劑摩爾分數單位的cmc,g表示反離子解離度,其他符號有其通常的含義。由于二甘醇是一種有機添加劑,因此膠束化的吉布斯自由能主要由表面活性劑-表面活性劑、表面活性劑-添加劑和添加劑-添加劑相互作用組成。Evans和Ninham[57]在考慮疏水和親水相互作用的同時,提出了以下方程式:

ΔGHP 0是將表面活性劑烴鏈從本體帶到膠束所需的能量的一部分,稱為膠束化的標準吉布斯能量,ΔGS0表示電荷頭和反離子之間發生的靜電相互作用。
在迄今為止研究的所有情況下,無論是在純水中還是在二甘醇-水二元混合物中,我們觀察到電導在cmc前后以不同的速率增加。在cmc以下,兩親性分子是非締合的,表現為1:1電解質,并遵循Debye-Huckel-Onsager方程[58,59],而在cmc以上,表面活性劑分子聚集并形成膠束[58,60]。cmc以上純CET、[C10min][Cl]和CET+[C10min][Cl]混合物的電導率急劇下降是由于膠束的形成導致材料的質量急劇增加,表面電荷密度凈降低。根據電導率與摩爾濃度圖中膠束后區域(S2)與膠束前區域(S1)中兩條相交直線的斜率之比計算g值,并可使用公式(10)[61]計算:

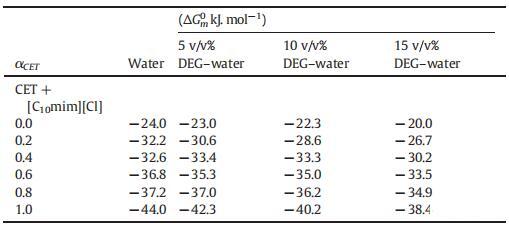
從表2中可以清楚地看出,g值并不遵循特定的趨勢,但是隨著DEG在混合系統中的體積百分比的增加,其g值以某種方式增加,這種增加可能是因為膠束的表面電荷密度降低,這是由于膠束聚集數減少所致。水和DEG-水二元混合物中CET、[C10min][Cl]和CET+[C10min][Cl]混合膠束的標準吉布斯自由能(ΔGm0)值如表3所示。如表3所示,隨著二甘醇與水的體積百分比的增加,膠束化過程變得不那么自發。
表2在T=298.15 K時,CET+[C10min][Cl]混合物在純水中以及在不同體積百分比的DEG-水二元混合物中的反離子離解度(g)值。
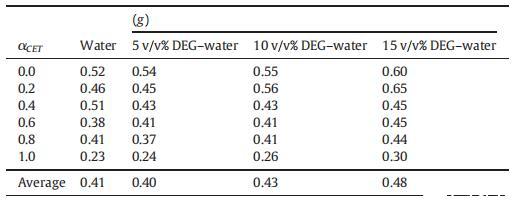
表3:在T=298.15 K時,CET+[C10min][Cl]混合物在純水以及不同體積百分比的二甘醇-水二元混合物中的膠束化標準吉布斯自由能(ΔGm0)值。
3.2.界面吸附
使用表面張力測量法測定了CET、[C10min][Cl]和混合體系CET+[C10min][Cl]在水和DEG-水混合溶劑中的表面活性(圖2)。表面張力值隨表面活性劑濃度的增加而減小,在某一特定區域,表面活性劑分子在氣液界面上的吸附接近于一個常數。表面張力測量對于計算這些表面活性劑及其混合體系的各種參數非常有用。
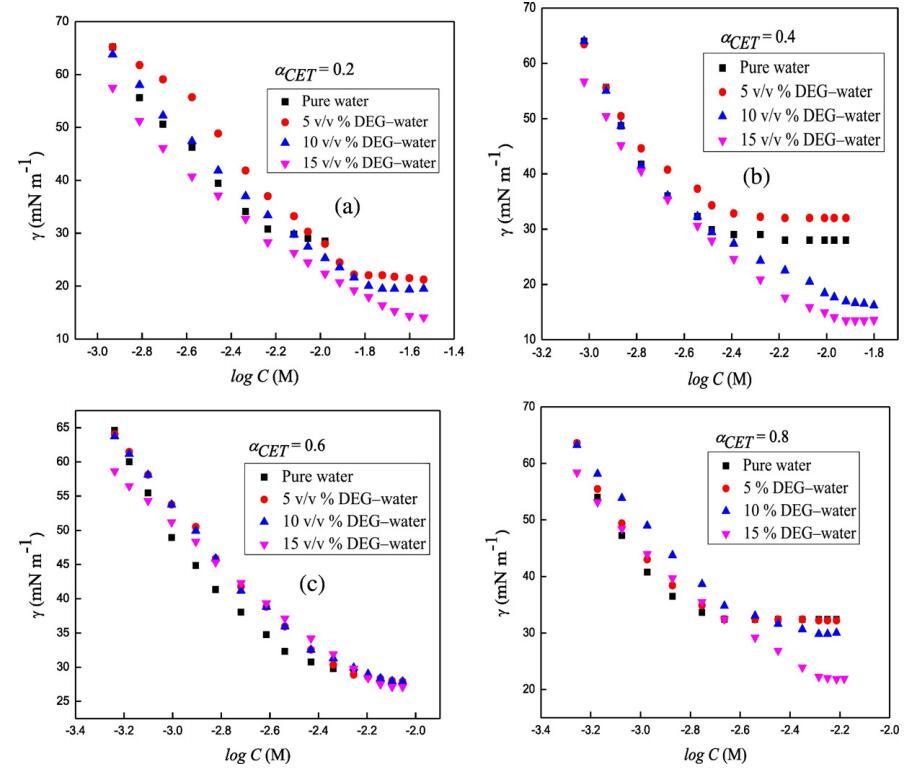
圖2.在T=298.15 K時,二甘醇-水二元混合物的不同體積百分比下,混合體系的表面張力,γ與對數[濃度]的關系曲線,CET+[C10min][Cl](a)αCET=0.2(b)αCET=0.4,(c)αCET=0.6和(d)αCET=0.8。
借助吉布斯吸附方程,我們可以使用公式(11)[62]計算表面活性劑在空氣-溶液界面的最大吸附量:
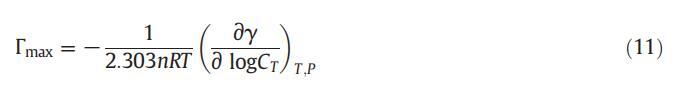
式中,γ是表面張力,n是溶液中由于表面活性劑和ILs離解而形成的物種數,CT是表面活性劑(兩親性)濃度,R是通用氣體常數,e?γ?logCTTT;P由恒定T和P下γ與對數CT的斜率得出。對于CET,n值為2,對于IL,n值為2,對于二元混合物,n值為4。從圖2中,我們可以通過γ和log C之間的線性圖計算斜率,用于計算表面過剩(Γmax),如公式(11)所示。
可用公式(12)[63]計算每個分子的最小面積:

其中NA是阿伏伽德羅的數字。表面過剩(Γmax)衡量表面活性劑在空氣/溶液界面的吸附效果,并取決于相互作用物種的分子結構。Γmax和Amin是描述表面活性劑分子在氣液界面上排列的非常重要的參數,也給出了表面活性劑及其二元混合物的發泡、潤濕和乳化特性的信息。通過比較Γmax值,我們發現Γmax按以下順序減小:Γmax(純)NΓmax(5 v/v%度)NΓmax(10 v/v%度)NΓmax(15 v/v%度)。此外,還觀察到,與混合系統相比,純系統的γ隨log C的下降速率更大,并且隨著混合系統中DEG體積百分比的增加,γ的下降速率進一步降低。然而,Amin隨著Γmax的增加而減少,其原因可能是界面上表面活性劑的數量減少導致Amin值增加。此外,當我們增加介質中DEG的體積百分比時,它與界面處的混合表面活性劑系統競爭,因此減少了其數量,這可以通過觀察到的Γmax和Amin值的趨勢清楚地顯示出來。與純水中的這些值相比,存在不同體積百分比DEG時不同摩爾分數CET(αCET)的Γmax值較低,原因可能是界面處存在非水介質,為混合表面活性劑系統提供的空間較小。不同摩爾分數的西替利胺(αCET=0.2、0.4、0.6和0.8)的Γ最大值為2.5×10?6,2.6×10?6,2.8×10?6和2.9×10?6摩爾米?2然而,在純水中,Γmax值隨著DEG量從5增加到15 v/v%而減小,在DEG的15 v/v%時,不同CET摩爾分數下的Γmax對應值為1.7×10?6,1.8×10?6,1.9×10?6和2.0×10?6摩爾米?分別為2。因此,Γmax值在存在15 v/v%的DEG時減少約32%,因此,這些混合系統在15 v/v%的DEG中的表面布居減少至約68%。不同CET摩爾分數(αCET=0.2、0.4、0.6和0.8)在純水中的Amin值分別為0.64、0.62、0.58和0.56 nm2,而DEG的15 v/v%中的相應值分別為0.89、0.88、0.86和0.78 nm2,因此Amin值增加了29.25%。
為了計算吸附效率,重要的是要知道產生最大吸附量的兩親分子的摩爾濃度。最大吸附量出現在表面張力降低到20mn·m時?1[64]和C20是該點的摩爾濃度。用pC20表示的兩親分子的吸附效率如下所示:

如表4所示,與二甘醇-水二元溶劑中的pC20值相比,純水中混合系統的pC20值更大,表明這些系統在純水中的吸附效率更好。
表4純CET、[C10min][Cl]和CET+[C10min][Cl]混合物的表面過剩(Γmax)、每分子最小面積(Amin)、吸附效率(pC20)和吸附自由能(ΔGad 0),作為純水中CET摩爾分數α的函數,并且在T=298.15K時存在不同體積百分比的二元DEG-水混合物。
吸附的標準吉布斯自由能可使用公式(14)計算:

式(14)中的術語e∏cmcΓmaxT為我們提供了將單層表面壓力為零的兩親分子引入膠束所需的工作信息。ΔGm0和ΔGad 0的值均為負值,表明這兩個過程都是有利的,但是ΔGad 0的值比ΔGm0的值更為負,這表明所有這些系統的吸附過程比膠束形成過程更為有利。
3.3.膠束中的堆積參數
純兩親分子及其混合體系的填充以及幾何結構可通過使用P表示的填充參數進行預測,并由以下關系式給出:

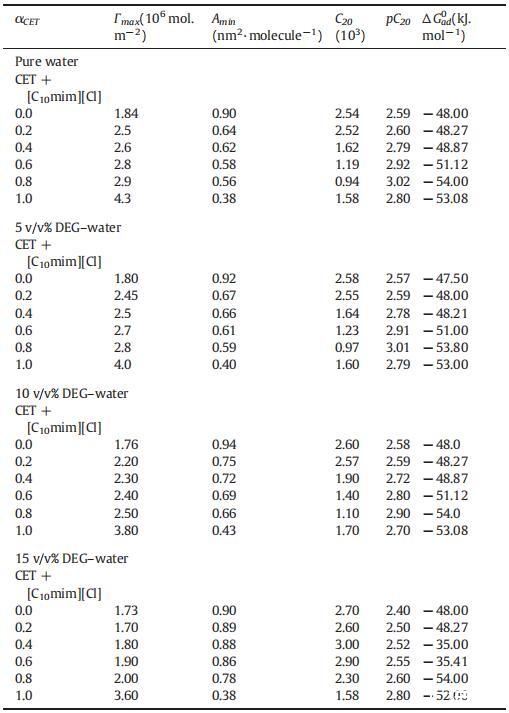
式中,v是視為流體且不可壓縮的疏水鏈的體積,lc是單體疏水鏈的長度,可使用Tanford公式計算[53]:
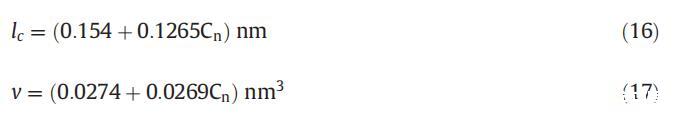
式中,Cn是烴鏈中碳原子的數量。對于混合膠束v,使用以下方程式計算:

其中i指第i種,xiR指根據魯賓方程計算的膠束摩爾分數,數值如表5所示。如果混合物為lCETNlIL,則考慮尾部長度較高的成分。純體系和混合體系的P值如表5所示。一般而言,對于球形體,P b 0.333,對于非球形體,P b 0.50,對于雙層和囊泡,P b 1為0.5,對于倒置結構,P N 1為。如表5所示,純體系及其混合體系的P值為b0。333,表明形成的膠束/混合膠束本質上是球形的。
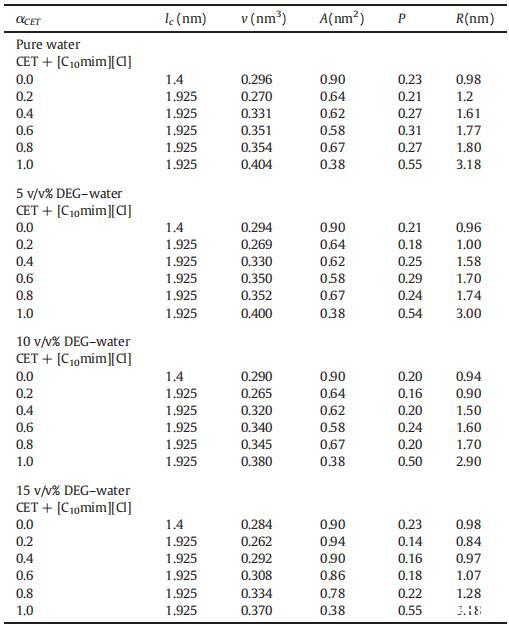
表5在純水中以及在T=298.15 K下存在不同體積百分比的DEG+水二元混合物時,純CET、[C10min][Cl]和混合物CET+[C10min][Cl]的有效疏水鏈長度(lc)、疏水鏈體積(v)、極性頭基團表面積(A)、填充參數(P)和半徑(R)的值。
3.4.紫外-可見光譜法研究IL-表面活性劑相互作用
圖5顯示了3×10的吸收光譜?5摩爾千克?1甲酚紅(CR)在純水中,吸收帶,即λmax出現在434 nm處,該值與其他地方報道的文獻值非常匹配[65]。離子表面活性劑表現出一些不同的聚集行為,即使在存在相對少量的帶相反電荷的染料的情況下。如圖5所示,吸光度先降低后升高,吸光度的降低可歸因于CET和染料分子之間形成的離子對,這些CET染料離子對也具有表面活性,CET濃度的進一步增加導致形成富染料膠束。隨著表面活性劑濃度的增加,這些富含染料的膠束轉化為表面活性劑膠束,當表面活性劑濃度達到其cmc值時,染料溶解[66]。然而,表面活性劑在CR中的cmc低于在純水中的cmc,這可能是由于帶正電的表面活性劑分子的頭基團之間的靜電斥力減少,如SO3?CR基團更喜歡與帶相反電荷的陽離子頭基團結合[65]。在將CET添加到IL+染料溶液中后,即使CET濃度低于純水中的濃度,也會形成cmc,原因可能是CET/IL混合膠束與可溶性染料分子的形成(圖6)。這里再次形成CET/染料和CET/IL離子對,但一旦CET達到其cmc值,它們都會分解為CET/IL混合膠束。圖7顯示了在二甘醇-水二元溶液存在下CET/IL混合膠束的吸收光譜。從圖7可以清楚地看出,隨著DEG體積百分比的增加,λmax向較低波長有一點偏移,即發生藍移,原因可能是溶劑極性降低。Ghanadzadeh等人[67]還表明,隨著溶劑極性的降低,結晶紫的光譜發生藍移。
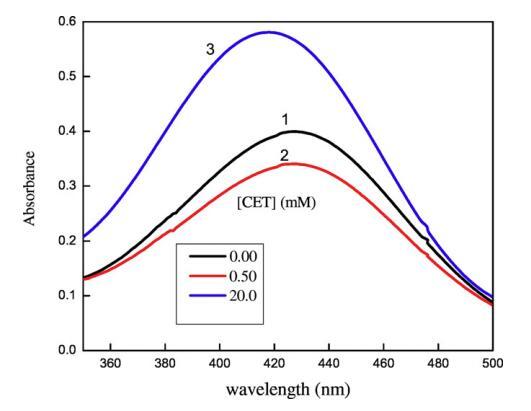
圖5.[CET]對水性甲酚紅染料可見光譜的影響:(1)[CET]=0mm,(2)[CET]=0.50mm,(3)[CET]=20mm。[CR]=3×10?5毫米。
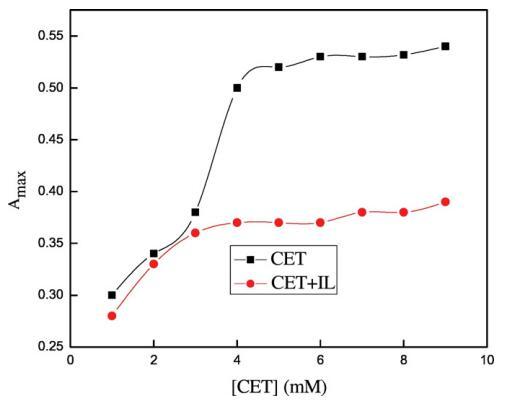
圖6.含水鉻峰的最大吸光度與[CET]的變化(■)在沒有ILs和()的情況下,存在0.05 w/w%的[C10min][Cl]。

圖7.含有不同體積百分比二甘醇-水混合物(1)5 v/v%二甘醇-水(2)10 v/v%二甘醇-水和(3)15 v/v%二甘醇-水的CET/[C10min][Cl]膠束聚集體中含水鉻吸收光譜的變化。